正文
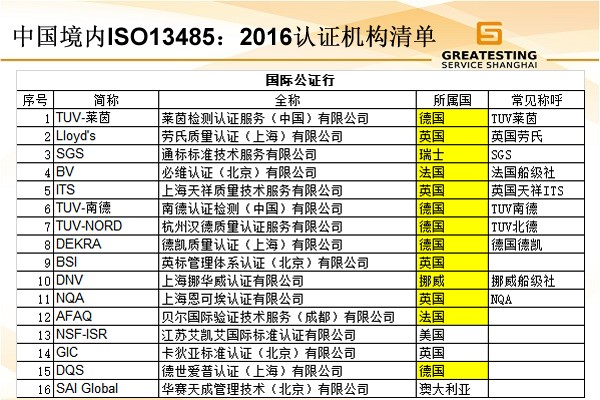
ISO13485医疗器械质量管理体系认证

本检测中心所获中国国家实验室认可委员会(CNAS)的实验室资质认可被全球55个国家互认,包括欧盟全境
本检测中心获得了美国消费品安全委员会(CPSC)颁发的实验室资质认可

本检测中心获得了美国消费品安全委员会(CPSC)颁发的实验室资质认可
ISO13485中文叫《医疗器械 质量管理体系 – 用于法规的要求》,当前的版本是ISO13485:2016版(于2016年3月1日颁布),对应的国家标准是YY/T0287-2017(于2017年1月19日发布)。ISO13485起源于ISO9001,开始时是作为ISO9001在医疗器械行业法规条件下的专用要求,但在2003年发布第二版之后,开始独立于ISO9001并作为单独的体系标准使用。
由于医疗器械是治病救人的特殊产品,产品固有的风险涉及的是人命关天的大事,为此各国均建立了各自的医疗器械GMP法规,而标准本身更多关注的也是法律法规和产品风险。2016版13485全文共提到了68处法规要求,每一处的法规要求都可能找到各国对应的法规要求,从而建立起ISO13485管理体系和各国法规的链接。所以建立并保持ISO13485质量管理体系的关键是识别法规要求,并将这些要求融入到企业的具体规章制度中去。
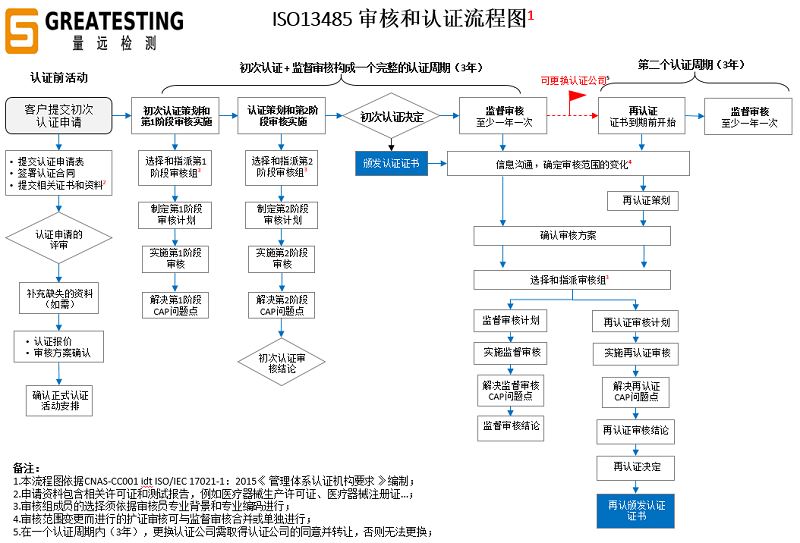
ISO13485咨询服务和认证流程:
第一步:联系我们并签订《咨询服务协议》;
第二步:联系认证公司,填写《审核申请表》并签署《认证合同》,提交相关资料;
需要预先提交的相关资料主要是:
1)营业执照副本复印件
2)产品第三方测试报告、生物相容性测试报告
3)食药监颁发的医疗器械生产许可证、注册证、登记表等
第三步:建立ISO13485质量管理体系并运行3个月;
第四步:进行第一阶段审核并完成问题点改善;
第五步:进行第二阶段审核并完成问题点改善;
第六步:颁发ISO13485证书;
第七步:进行一年一次的监督审核并完成问题点改善(至少2次);
第八步:证书到期前,进行再认证审核并完成问题点改善(第二个认证周期)
ISO13485体系申请条件:
1)营业执照满3个月
2)生产型企业
3)食药监颁发的相关证件证书
4)建立相应的洁净车间(按法规要求)
ISO13485证书办理需多长时间:
1)简单辅导型:单体系1个月左右,双体系1.5半个左右,三体系2个月左右,具体以各认证机构审核安排为准。
2)系统培训型:如果需要好好做,导入培训,系统辅导,周期大概是3个月左右。
3)以上时间未包含认证机构要求的第1阶段和第2阶段的时间间隔(大部分机构在1个月左右),证书批准时间和制证时间(各机构差异较大)
3)以上时间未包含认证机构要求的第1阶段和第2阶段的时间间隔(大部分机构在1个月左右),证书批准时间和制证时间(各机构差异较大)
ISO13485证书有效期几年:
1)证书有效期均是3年。
ISO13485是否要年审:
1)每年要进行监督审核。
ISO13485认证对企业有什么好处:
1)提高企业遵守《医疗器械生产质量管理规范》的能力,帮助企业顺利获得和保持《医疗器械生产许可证》;
2)提高企业遵守各国医疗器械GMP法规的能力,例如欧盟MDD/MDR & IVDR法规、美国 FDA QSR 820法规、日本J-Pal法规169、韩国KGMP法规、巴西BPF法规等;
3)规避法律风险, 通过有效的风险管理,有效降低产品出现质量事故或不良事件的风险;
4)提高和改善企业的管理水平,增加企业的知名度;
5)提高和保证产品的质量水平,使企业获取更大的经济效益;
6)有利于消除贸易壁垒,取得进入国际市场的通行证;
7)有利于增强产品的竞争力,提高产品的市场占有率。

目前大多数医疗设备生产厂家在建立质量管理体系时,开始把ISO9001+ISO13485+CE认证作为一揽子解决方案来考虑。下面我们就来解释欧盟医疗器械CE认证与ISO13485的关系:
一、进入欧盟医疗器械市场到底需不需要进行ISO13485:2016体系认证?
答:在我们的日常咨询工作中经常碰到客户的这个问题,有些人回答需要,有些人回答不需要。尤其是新冠疫情爆发以来,很多防疫物资厂商在这个问题上疑惑不解,那么到底需不需要?下面笔者以医用口罩为例为大家讲解说明:
1)非灭菌医用口罩(不需要) – 只需要根据EN14683产品标准完成产品测试、CE技术文档、自我符合性声明和欧代注册就可以合法在欧盟上市。
2)灭菌医用口罩(需要) – 除了非灭菌口罩的步骤要求以外,还额外需要进行质量管理体系审核并取得公告机构(第三方)颁发的ISO13485证书和CE证书。
而对于其他产品而言更能面临更为复杂多变的符合性评估流程,所以到底要不要进行ISO13485:2016的认证需要根据产品的具体分类和法规细节要求进行,我们不能简单的一句说某个产品需要,某个产品不需要。
二、那么为什么会出现这样的差异?
这是由于欧盟的医疗器械分类导致的,当前欧盟的MDR法规列举了22条规则对医疗器械进行分类,分类方法复杂而细致。分类规则的核心依据是产品的预期用途和固有风险进行,但是根据预期使用目的的不同,同一个产品的分类千差万别,厂商在编制《产品使用说明书》时需要谨慎对待。例如电热褥,根据其使用目的的不同,可以做如下分类:
1. 非医疗器械(日常家居用品);
2. I类医疗器械(规则1:非侵入式医疗器械);
3. IIa类医疗器械(规则9:手术电热衣褥)
4. IIb类医疗器械(规则9:临床新生儿保温)。
上面提到的医用口罩在分类中属于I类医疗器械,根据MDR第V章第2节第52条“符合性评估流程”的规定,I类器械中“无菌提供”或含“测定功能”还是需要进行管理体系审核。很明显灭菌医用口罩需要公告机构(第三方)介入并进行认证审核。当然II类和III类肯定是需要由公告机构进行认证的。
三、医疗器械CE认证必须采用ISO13485:2016吗?
除了部分I类医疗器械只需要进行自我符合性声明,大部分医疗器械的CE认证必须按照ISO13485:2016的规定建立质量管理体系,CE认证的现场审核主要依据13485、欧盟MDD/MDR法规和相关产品标准进行。在最新公布的(EU)2020/437《MDD医疗器械协调标准决议》的清单中列举了欧盟医疗器械的所有相关协调标准,ISO13485:2016是唯一的质量管理体系标准,这也让ISO13485成为欧盟医疗器械CE认证的强制标准。
部分法规依据如下:
1)欧盟医疗器械的CE认证根据(EU)2017/745 《MDR欧盟医疗器械法规》(以下简称MDR法规)进行,从2020年5月26日开始执行。由于新冠疫情的原因,MDR法规执行推迟至2021年5月26日,当前执行的仍是93/42/EEC《MDD欧盟医疗器械法规》(以下简称MDD法规)。
2)依据MDR法规第V章第1节第51条“器械分类”的规定,欧盟的医疗器械分为四类(具体分类依据参照MDR法规“附录VIII分类规则”列举的22条分类规则)。
根据器械的预期用途和其固有风险,医疗器械应分为I、IIa、IIb和III类。分类应按照附录VIII规定进行。
3)依据(EU)2017/745 MDR欧盟医疗器械法规第V章第2节第52条“符合性评估流程”的规定,IIa、IIb和III类和部分I类需要进行符合性评估和管理体系审核(具体的评估流程见附录IX“基于质量管理体系和技术文件评估的符合性评估”)。需要注意对I类器械的如下规定:
I类器械制造商...规定的技术文件后,须通过签发...EU符合性声明,以声明其产品的符合性。若这些器械在无菌状态下投放市场,具有测定功能或为可重复使用手术器械,制造商应采用附录IX第I章和第III章或附录XI第A部分所述程序。但公告机构的介入应限于:
(a) 若此类器械在无菌状态下投放市场,则涉及建立、保障和保持无菌条件。
(b) 若此类器械具有测定功能,则涉及器械符合计量要求的情况。
4)依据(EU)2017/745 MDR欧盟医疗器械法规第II章第8条“使用协调标准”的规定,进入欧盟市场的医疗器械医药采用与其官方公报上的协调标准相一致
本法规引用的协调标准参考资料应理解为与发表在欧盟官方公报上的参考资料一致。
5)根据2020年3月25日公布的(EU)2020/437《MDD医疗器械协调标准决议》,附表I(第120行)和附表II(第6行)仍延续之前规定将ISO13485:2016作为欧盟医疗器械CE认证的管理体系协调标准
延伸阅读(点击了解详情):

上一篇:沃尔玛验厂突击审核
下一篇:暂无
验厂验货
联系我们
电话:021-64788791
邮箱:info@greatesting.com
地址:上海市闵行区莲花南路2899弄莲谷科技园1号楼10楼
 Jerry Qin
Jerry Qin